1. Introduction
Fibrodysplasia ossificans progressiva (FOP) is a rare but devastating autosomal dominant genetic disorder characterized by spontaneous or trauma-induced progressive extra-skeletal bone formation, called heterotopic ossification (HO), in skeletal muscles, tendons, and ligaments [1]. HO forms through endochondral ossification, the process that creates most bones during embryonic development [2]. Endochondral ossification occurs through the formation of a cartilaginous structure followed by cartilage resorption and its replacement by bone [2]. In the case of FOP, endochondral osteogenesis occurs extra-skeletally, replacing soft connective tissues with bone tissue [1]. Causative missense mutations for FOP occur in the ACVR1 gene, which encodes a type I bone morphogenic protein (BMP) receptor [3], with ACVR1R206H being the most prominent mutation, occurring in an estimated 97% of FOP patients [4,5,6,7]. The prevalence of FOP has been estimated to be approximately 0.6–1.39 per million inhabitants, with around 900 confirmed cases worldwide [1,8,9,10]. No sex, racial, or ethnic patterns have been observed in FOP patients [1,8,9,11,12,13]; however, not enough populations have been investigated to conclusively exclude these confounding factors. Around half of FOP patients are misdiagnosed, most commonly with cancer, which has often lead to a 5–6-year delay in the correct diagnosis [11,12,13,14]. While misdiagnoses are common in many rare diseases, increasing FOP awareness within the medical community and globally is necessary to lessen preventable harm to patients due to intramuscular injections, invasive biopsies, and surgical procedures [14,15].
Most patients with FOP are born with congenital malformation of the great toe, but otherwise appear normal at birth [1]. Children with FOP start experiencing unpredictable episodes of painful soft tissue swelling, known as flare-ups, around the age of 5, but onset varies per individual [6,11,12,16]. Flare-ups often lead to irreversible HO along the body and joints; however, ectopic bone formation has also been reported in the absence of flare-ups [6,11,12,16]. Even though flare-ups and new episodes of HO can be triggered by muscular trauma, they also occur spontaneously, supporting the critical role of the immune system in the pathogenesis of FOP. However, the cause of these local inflammatory symptoms remains unclear. Heterotopic bone forms more commonly in the upper body before age 8, while over time, HO formation occurs more distally and in the lower limbs [11,12,16]. Cumulative HO formation restricts range of motion, causes severe pain, and gradually immobilizes patients, reducing their quality of life. Most heterotopic bone forms during adolescence and young adulthood; with age, accumulation of new HO volume decreases, which may be attributed to reduced HO initiation and tissue availability [11]. Disease progression leads to most patients needing aids, assistive devices, and adaptations (AADAs) by their 20s [11]. Progressive HO shortens the lifespan of individuals with FOP, with 56 years as the median life expectancy [15]. The leading cause of death among patients is thoracic insufficiency syndrome, caused by cardiorespiratory failure due to ossification of the rib cage [15].
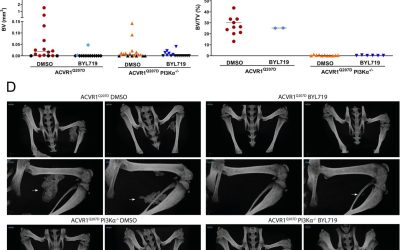
There is no current cure for FOP patients, though, in a significant advancement, Palovarotene, a selective retinoic acid receptor gamma (RARy) agonist, has been recently approved by the FDA in the United States (US) and by respective agencies in Canada and Australia as a treatment to reduce new HO formation [17,18,19,20]. However, concerns for skeletal growth were raised during clinical trials and a pre-clinical study [18,20,21], resulting in Palovarotene only being approved for patients aged ≥8 and ≥10 years for females and males, respectively in the US, Australia, and Canada [17,20]. Considering these advancements, research into FOP pathogenesis and the development of other effective treatments, some already in clinical trials [22], are necessary so children with FOP can be treated, given that significant HO progression occurs during childhood.
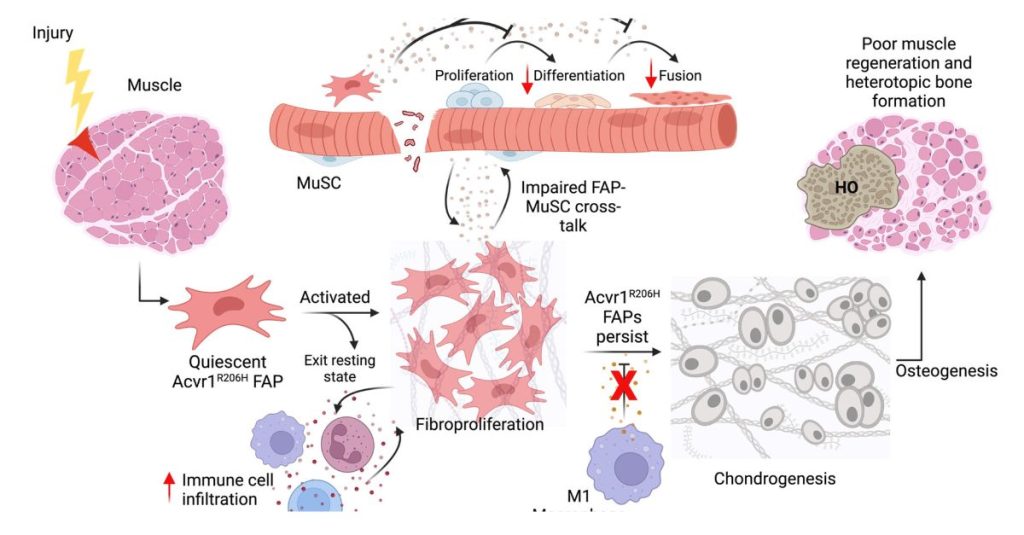
In the last few years, considerable progress in FOP research has led to the identification of cellular progenitor(s) of HO. Tissue-resident mesenchymal progenitors have been implicated as cellular origins of ectopic cartilage [23]. The development of treatments targeting these progenitors has potential to relieve the burden of HO in patients. In this review, we highlight the cellular sources of HO, focusing on the emerging role of fibro-adipogenic progenitors (FAPs) in the pathogenesis of FOP.
2. ACVR1-Mediated Signaling and FOP
ACVR1 is one of the cell surface receptors that mediate bone morphogenic protein (BMP) signaling [24,25,26]. BMP signaling has been extensively studied for its role in skeleton patterning, endochondral skeletal development, growth plate growth, and chondro-osteogenic differentiation [24]. ACVR1KO studies show the receptor’s crucial role in endochondral bone formation and craniofacial development, as well as non-bone processes such as cardiac development and neurogenesis [25]. BMPs also participate in a variety of biological processes such as cell proliferation, differentiation, embryogenesis, and development, as well as in adult tissue homeostasis in bone and other tissues [26]. ACVR1 signals through suppressor of mothers against decapentaplegic (SMAD)-dependent (canonical) and SMAD-independent (non-canonical) pathways [24,25,26]. This section will define the role of ACVR1 in bone regulation and homeostasis, including how dysregulated ACVR1R206H-receptor signaling drives FOP pathogenesis.
Figure 1 summarizes how canonical and noncanonical ACVR1-mediated signaling is altered in FOP. Specific pharmacological targeting of these pathways has been studied to ablate HO formation, with multiple preclinical studies showing promise for new FOP treatments.
Source: NCBI