DISEASE OVERVIEW
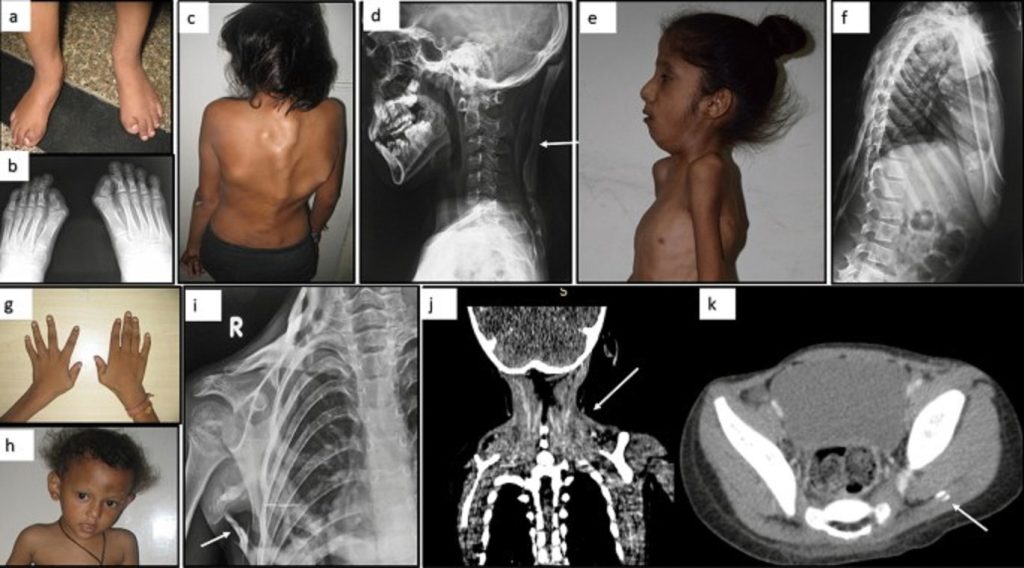
Fibrodysplasia ossificans progressiva (FOP) is an extremely rare genetic connective tissue disorder characterized by the abnormal development of bone in areas of the body where bone is not normally present (heterotopic ossification), such as the ligaments, tendons and skeletal muscles. Specifically, this disorder causes the body’s skeletal muscles and soft connective tissues to undergo a metamorphosis, essentially a transformation into bone, progressively locking joints in place and making movement difficult or impossible. Patients with FOP have malformed big toes that are present at birth (congenital). Other skeletal malformations may occur. The abnormal episodic development of bone at multiple soft tissue sites leads to stiffness in affected areas, limited movement and eventual fusion (ankylosis) of affected joints (neck, back, shoulders, elbows, hips knees, wrists, ankles, jaw – often in that order).
Episodic flare-ups (inflammatory soft tissue swellings) of FOP usually begin during early childhood and progress throughout life. Most cases of FOP occur as the result of a sporadic new variant in the ACVR1 gene in the bone morphogenetic protein (BMP) signaling pathway, which is important during the formation of the skeleton in the embryo and the repair of the skeleton following birth.
SYNONYMS
- FOP
- myositis ossificans progressiva
SIGNS & SYMPTOMS
All individuals with classic FOP have malformations of the great toes and, in approximately 50% of patients, the thumbs. These changes in the skeleton are present at birth (congenital) and are the first clinical signs of this disorder. The most common skeletal malformation associated with FOP is a shortened great toe with a malformed distal first metatarsal and a missing or abnormal first phalanx and/or interphalangeal joint. Other malformations of the toes and fingers may include inward turning of the great toe toward the other toes (hallux valgus), abnormally short fingers and toes (microdactyly) and/or permanent fixation of the fifth finger in a bent position (clinodactyly). Other congenital signs of FOP include proximal medial tibial osteochondromas, malformation of the upper part of the spinal column (cervical vertebrae) and an abnormally short broad neck of the bone in the thigh that extends from the knee to the pelvis (femur).
Progressive endochondral bone formation in connective tissues (heterotopic ossification) usually occurs during early childhood and progresses throughout life. Abnormal development of bone may occur spontaneously but often occurs following an episode of soft tissue injury or a viral illness. The first sign of heterotopic ossification is the appearance of firm tender swellings (often referred to as flare-ups) on certain parts of the body, especially the back, neck and/or shoulders. These soft tissue swellings mature through a cartilage-to-bone (endochondral) pathway to form mature heterotopic bone. The ectopic bone growth usually involves tendons, ligaments, skeletal muscle tissue and connective tissues such as fascia and aponeuroses. In many patients, pain and stiffness occurs in these areas. On some occasions, a low-grade fever may herald the development of these swellings. Although the swellings eventually regress, they usually harden into mature bone as they decrease in size.
In the affected areas, bone slowly replaces connective tissue. The neck, back, chest, arms and legs are usually the first areas affected. The disease eventually affects the hips, ankles, wrists, elbows, shoulders and/or jaw as well as the abdominal wall. In some affected individuals, the progression of bone development may be rapid; in others, the process may be gradual. Even among identical twins, the disease progression may vary greatly, reflecting different environmental impacts such as traumatic episodes.
Chronic swelling in various parts of the body is a common physical characteristic of individuals with FOP. Swelling may occur coordinately with the abnormal bone formation that characterizes FOP, or it may occur when recently formed bone presses on lymphatic vessels, obstructing the flow of tissue fluid. In addition, swelling may also be caused by a lack of pumping action within the hardened (ossified) muscle and can cause blood and tissue fluids to pool in a limb (e.g., arms and/or legs).
Abnormal development of bone eventually leads to stiffness and limited movement of affected joints. If the jaw is involved, the person may have trouble eating and/or speaking. In addition, abnormal development of bone may lead to progressive deformity of the spine including side-to-side (scoliosis) and, in some people, front-to-back curvature of the spine (kyphosis). As is the case for skeletal bone, the bone that develops in abnormal areas may fracture and then undergo fracture repair. As the disease progresses, individuals with FOP experience increasingly limited mobility that causes problems with balance, difficulty walking and/or sitting and/or severely restricted movement.
FOP may eventually result in complete immobilization. Affected individuals may have progressive pain and stiffness in affected areas, complete fusion of the spine and/or pain in affected areas of the body caused by abnormal bony growths that compress the nerves in these areas (entrapment neuropathies). As mobility begins to deteriorate, affected individuals may have an increased susceptibility to respiratory infection or right sided congestive heart failure due to thoracic insufficiency. Hearing impairment is seen in approximately 50% of affected individuals. Some people with more severe forms of FOP may have hair loss or mild cognitive delay.
CAUSES
Most cases of FOP occur sporadically, and there is a single affected individual within a family. When a familial pattern has been identified, FOP is inherited in an autosomal dominant pattern.
Dominant genetic disorders occur when only a single copy of a disease-causing gene variant is necessary to cause the disease. The gene variant can be inherited from either parent or can be the result of a new (de novo) changed gene in the affected individual that is not inherited. The risk of passing the gene variant from an affected parent to a child is 50% for each pregnancy. The risk is the same for males and females.
In 2006, an international team of researchers led by Eileen M. Shore, PhD and Frederick Kaplan MD at the University of Pennsylvania, published results of research identifying the genetic cause of FOP. The team found that FOP is caused by a change (variant) in the ACVR1 gene in the bone morphogenetic protein (BMP) signaling pathway.
Bone morphogenetic proteins are regulatory proteins important in embryonic skeletal formation and in post-natal repair of the skeleton. The ACVR1 encodes a BMP receptor called activin receptor type IA, or ACVR1, one of four known BMP type I receptors. BMP receptors, located at the cell surface, help determine the fate of the stem cells in which they are expressed by transmitting signals into the cell. The classic clinical FOP presentation is caused by the specific substitution of a particular amino acid (arginine, at position 206) in the ACVR1 protein for another amino acid (histidine). This amino acid substitution induces activation of signaling by the ACVR1 receptor.
AFFECTED POPULATIONS
FOP is a very rare inherited connective tissue disorder that was first identified in the 18th century. The prevalence of FOP is estimated to be 1/1,000,000. FOP affects males and females equally, and people from all ethnicities.
DISORDERS WITH SIMILAR SYMPTOMS
Symptoms of the following disorders may be similar to those of fibrodysplasia ossificans progressiva. Comparisons may be useful for differential diagnosis:
Aggressive juvenile fibromatosis is a condition in which cells called fibroblasts increase in number in tendons, ligaments and other connective tissues. This overgrowth of cells may invade adjacent tissues, causing pain and disability. The resulting lesions may resemble the tissue swelling associated with FOP. However, individuals with aggressive juvenile fibromatosis do not have the toe malformation that is associated with FOP, nor do they develop heterotopic ossification.
Progressive osseous heteroplasia (POH) is an extremely rare inherited disorder characterized by the abnormal development of bone in areas of the body where bone is not normally present (heterotopic ossification). Unlike FOP, the initial bone growth may develop on the surface of the skin (osseous plaques). These areas may become progressively widespread and come together (coalesce) to form even larger areas of hardened and thickened skin (dermal ossification). POH spreads to deeper levels of the skin and to various muscle, fatty and connective tissues of the body. As the disorder continues to progress, the abnormal development of bone may lead to stiffening and limited movement of affected joints. In severe cases, affected joints may become permanently fixed (ankylosed). In addition, arms and legs may become malformed and not grow to full length. POH is caused by a variant in the GNAS gene. GNAS variants have been identified in approximately 70% of affected individuals. POH is a distinct developmental disorder and belongs to a spectrum of clinical conditions that have the common feature of ossification of the skin (osteoma cutis). Although skeletal malformations have been noted, individuals with POH do not have the toe malformation that is characteristic of FOP. (For more information on this disorder, choose “Progressive Osseous Heteroplasia” as your search term in the Rare Disease Database.)
DIAGNOSIS
Misdiagnosis of FOP is common but can be avoided by examining the individual’s toes for the characteristic feature, short great toes. The diagnosis may be confirmed by a thorough clinical evaluation, characteristic physical findings, and sequencing of the ACVR1 gene.
STANDARD THERAPIES
Treatment
Biopsies should be avoided when FOP is suspected because these tests may result in rapid bone formation in those areas where tissue is removed. Intramuscular injections (e.g., immunizations) must be avoided. Injections of local anesthetics and stretching of the jaw for dental therapy should be avoided. People with FOP should avoid any situations, such as falls, that may cause blunt trauma, since trauma may cause abnormal bone development. Various viral illnesses including influenza and influenza-like illnesses may provoke flare-ups of the condition.
In 2023, palovarotene (Sohonos), a retinoic acid receptor γ (RARγ) agonist, was approved by the U.S. Food and Drug Administration (FDA) as the first treatment for FOP to reduce extra-skeletal bone formation in adults and children aged 8 years and older for females, and 10 years and older for males.
Preventative (prophylactic) antibiotic therapy may be appropriate to prevent infection in affected individuals with an increased susceptibility to respiratory infections due to progressive mobility impairment.
Certain types of drugs have been used to relieve pain and swelling associated with FOP during acute flare-ups (most notably corticosteroids) and non-steroidal anti-inflammatory medication between flare-ups.
Affected individuals may benefit from occupational therapy. Special shoes, braces and other devices that assist in walking and weight-bearing have been used to help people with FOP. An occupational therapist can help obtain special devices or tools to assist in daily activities.
A team approach for infants diagnosed with FOP is also recommended and may include special social, educational and medical services.
Genetic counseling is recommended for individuals with FOP and their families. Other treatment is symptomatic and supportive.
Recent Posts
-
 A Week of Preventive Treatment and Emergency Flare-Up Management
A Week of Preventive Treatment and Emergency Flare-Up Management -
 Mayeshree’s Journey — Staying Ahead of Future Flare-Ups
Mayeshree’s Journey — Staying Ahead of Future Flare-Ups -
 Coordinating Hospitals and Doctors Across India for Seamless FOP Care
Coordinating Hospitals and Doctors Across India for Seamless FOP Care -
 FOP Awareness Expands — Training Program Held for Police Personnel
FOP Awareness Expands — Training Program Held for Police Personnel -
 Global Guidelines Continue to Shape Local Treatment Decisions
Global Guidelines Continue to Shape Local Treatment Decisions