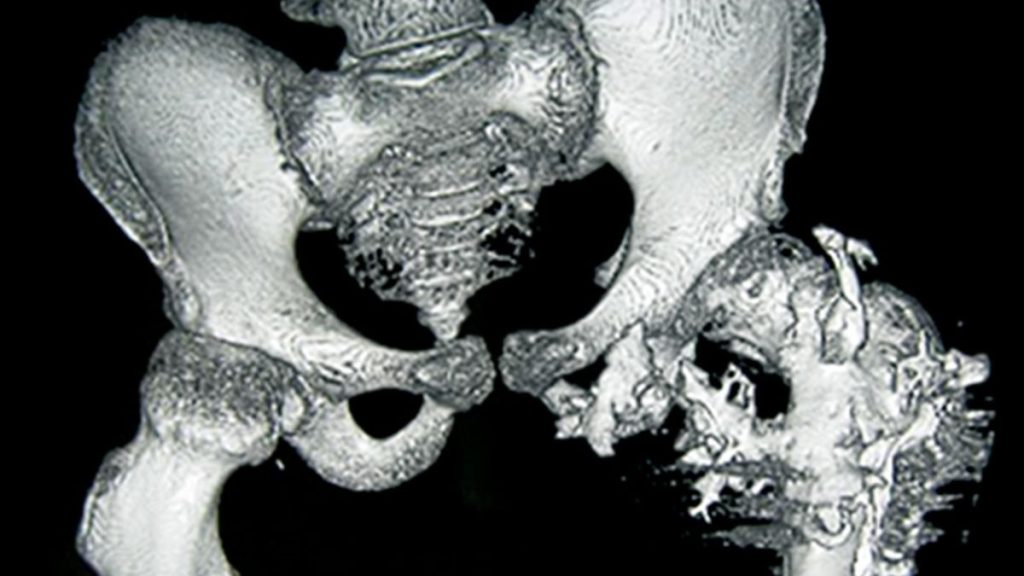
Rare diseases often get the cold shoulder from drug developers, and fibrodysplasia ossificans progressiva (FOP), an infamous genetic condition that makes bone sprout where it shouldn’t, is one of the rarest. It can inflict terrible pain and disability and is ultimately fatal, but only affects some 8000 people around the world.
Yet at least 13 companies are chasing therapies for FOP, and all this effort has started to pay off. Last year, the U.S. Food and Drug Administration approved the first drug to treat the disease, palovarotene. Four others are in trials, including a compound described this week in Science Translational Medicine that stymies the defective protein behind the disease.
Thanks to this progress, scientists and doctors say, a suite of drugs could soon be available to treat the fatal condition. They’re likely to be costly because the market is so small; palovarotene runs $624,000 per year in the United States. But the compounds may stop progression of the disease and could even allow surgeons to remove out-of-place bone without exacerbating the illness. “I hope we can turn a catastrophic disease into an inconvenience,” says Frederick Kaplan, an orthopedic surgeon at the University of Pennsylvania (UPenn) Perelman School of Medicine who has been studying FOP for more than 30 years.
Kaplan and other researchers suggest one reason drug developers have focused on FOP is the cruelty of the disease, which is triggered by a mutation in a gene crucial to bone growth. Children with FOP usually seem fine at birth, but as they get older bone starts to form in their muscles, tendons, and ligaments. The frequent falls and bruises of childhood can spark these spates of bone development, as can surgery and even injections—forcing patients to skip vaccinations.
As the disease progresses, the superfluous bone can contort the body and lock up joints, making it difficult to walk and eat. Patients are usually in wheelchairs by age 30 and die in their 50s, often because the excess bone hampers breathing. After seeing the suffering, “researchers feel obligated to do something,” says developmental biologist Maurizio Pacifici of the Children’s Hospital of Philadelphia, whose lab identified palovarotene as a potential FOP treatment.
Researchers also hope the disease will provide insights into how to treat more common conditions in which bone formation goes awry. For example, people without FOP sometimes accumulate misplaced bone after burns, blast injuries, and hip replacements, or when they develop heart disease.
Not that long ago it wasn’t even clear the disease was genetic, says Eileen Shore, a geneticist and cell biologist also at UPenn, which is the epicenter for studies of the disease. “We couldn’t get pharma [companies] interested,” she recalls.
But when she, Kaplan, and their colleagues identified the faulty gene behind FOP in 2006, “that changed everything,” Shore says. The disease results from mutations in the gene for ALK2, a surface protein on many kinds of cells, including certain stem cells. Researchers don’t fully understand the disease mechanism, but the abnormal ALK2s are overactive. They may dispatch messages that cause stem cells to specialize, triggering skeletal eruptions at the wrong time and place.
That molecular pathway revealed possible drug targets. Instead of taking aim at ALK2, Pacifici and his colleagues decided to interdict the pathway farther downstream by activating a molecule that blocks formation of cartilage, bone’s precursor. The researchers tested several related molecules, including palovarotene, which had the advantage of already being in clinical trials for another condition. A 2016 study showed that the drug curbs new bone buildup in mice genetically engineered to mimic FOP.
Palovarotene seems to do the same in people. In a phase 3 trial reported last year, patients who received the drug accrued 60% less new bone per year than patients in a separate study who received no therapy.
As the first approved targeted treatment for FOP, palovarotene became a landmark. But it has several drawbacks besides its cost. It doesn’t stop the disease. Moreover, young children can’t take it for fear it will stunt their growth, so it isn’t used to combat the early stages of the illness, Kaplan says. “The battleground in FOP is children.”
So far, only four countries—the U.S., Canada, Australia, and the United Arab Emirates—have approved palovarotene, which is made by the French pharma company Ipsen. “It’s exciting that there is a drug, but the reality is we can do better with targeted treatments,” says endocrinologist Marc Wein of Massachusetts General Hospital, who is not involved in developing the drug.
Researchers at the pharmaceutical company Regeneron have devised a monoclonal antibody, garetosmab, that may overcome some of palovarotene’s limitations but has brought some new concerns. In 2015, researchers at the company and an independent group in Japan discovered that the faulty version of ALK2 becomes overactive when it is stimulated by a protein called activin A, to which normal ALK2 doesn’t respond. Garetosmab clamps onto activin A, blocking the abnormal stimulation.
A phase 2 trial of garetosmab in 44 patients with FOP, published in 2023 in Nature Medicine, showed that although 41% of one patient subgroup sprouted additional bone lesions during 7 months on a placebo, only 5% did after they began receiving garetosmab. Regeneron recently began recruiting patients for a phase 3 trial to further test the drug’s effectiveness—and safety.
The latter is a major issue as five patients died in the garetosmab phase 2 trial. “We looked under every stone we could to understand what happened to these patients,” says Dushyanth Srinivasan, medical director of the garetosmab program at Regeneron. Participants in the trial were ill, and in the end, the consensus was that the drug did not cause the deaths, he says. But Kaplan, who was a co-author on the Nature Medicine paper, says the safety of garetosmab remains a concern. Activin A performs a variety of jobs in the body, so blocking it may lead to side effects.
Three other drugs in clinical trials take aim at ALK2 itself. The protein is a kinase, an enzyme that turns other proteins on or off by affixing phosphate molecules to them, and the new compounds jam its active site. One, developed by Alison Davis, then a lead biologist at Blueprint Medicines, and colleagues, halted formation of new bone in mice with an FOP-like condition, the researchers report this week in Science Translational Medicine. Now owned by Ipsen and dubbed fidrisertib, the drug is already in a phase 2 trial. The other two ALK2 inhibitors are also in phase 2 trials.
Although the kinase inhibitors are promising, blocking ALK2 could also trigger side effects, notes medical geneticist Toshifumi Yokota of the University of Alberta. (FOP patients still have one normal gene for the protein.) He has helped start a company to probe whether oligonucleotides, short strands made of the building blocks of RNA or DNA, can inhibit the protein more selectively.
Even if the new drugs only stop the disease rather than reversing it, they could be a major help. Surgeons hesitate to cut away excess bone in patients with FOP because the tissue trauma spurs yet more bone formation. “If we could prevent reformation of bone after surgical removal, that would be a coup,” Pacifici says.
This surge in drug development is encouraging for patients with FOP, Wein says. But he adds that it should also be heartening for patients with other rare illnesses, who should think, “if they can do it for FOP, they can do it for my disease.”
Recent Posts
-
 A Week of Preventive Treatment and Emergency Flare-Up Management
A Week of Preventive Treatment and Emergency Flare-Up Management -
 Mayeshree’s Journey — Staying Ahead of Future Flare-Ups
Mayeshree’s Journey — Staying Ahead of Future Flare-Ups -
 Coordinating Hospitals and Doctors Across India for Seamless FOP Care
Coordinating Hospitals and Doctors Across India for Seamless FOP Care -
 FOP Awareness Expands — Training Program Held for Police Personnel
FOP Awareness Expands — Training Program Held for Police Personnel -
 Global Guidelines Continue to Shape Local Treatment Decisions
Global Guidelines Continue to Shape Local Treatment Decisions