Introduction
Fibrodysplasia ossificans progressiva (FOP) is a rare, progressively debilitating disorder first described in 1692 by Guy Patin [1]. This disease is characterized by progressive heterotopic ossification that forms normal bone in characteristic extraskeletal sites and congenital malformations of the great toes. Progressive episodes of heterotopic ossification occur in characteristic pattern first at the dorsal, axial, cranial, and proximal part of the body and later in the ventral, appendicular, caudal, and distal part [2]. The heterotopic ossification contributes to a severe restriction of movement, progressing to total immobility, and ultimately leading to mortality from cardiorespiratory complications often by the fourth decade of life [3]. The incidence of FOP is 1 in 2 million individuals, with no observed preference for gender, race, or ethnicity [4].
Most FOP patients have a spontaneous new mutation in the ACVR1 gene, which encodes the bone morphogenic protein type 1 receptor [5]. The mutation results in a form of the receptor that’s overly active, even in the absence of its ligand. This results in abnormal activation of osteogenesis at ectopic sites [6]. Though familial cases have been reported, most FOP cases are sporadic [7].
While the genetic cause of FOP is now well established, the clinical course of the disease can vary greatly among patients. An international, natural history study has helped shed light on the debilitating effects and progressive nature of FOP, with the greatest progression noted during childhood and early adulthood [8]. Despite this, the rarity of FOP and its diverse clinical manifestations, as illustrated by a case series reported by Chan et al. in Hong Kong, can result in delayed or incorrect diagnoses [9].
Awareness of FOP among practitioners across a broad spectrum of specialties is key for early diagnosis. Early diagnosis not only slows progression but can help avoid potentially harmful procedures, such as biopsies or surgical interventions, which can exacerbate the condition [10,11].
Case 1
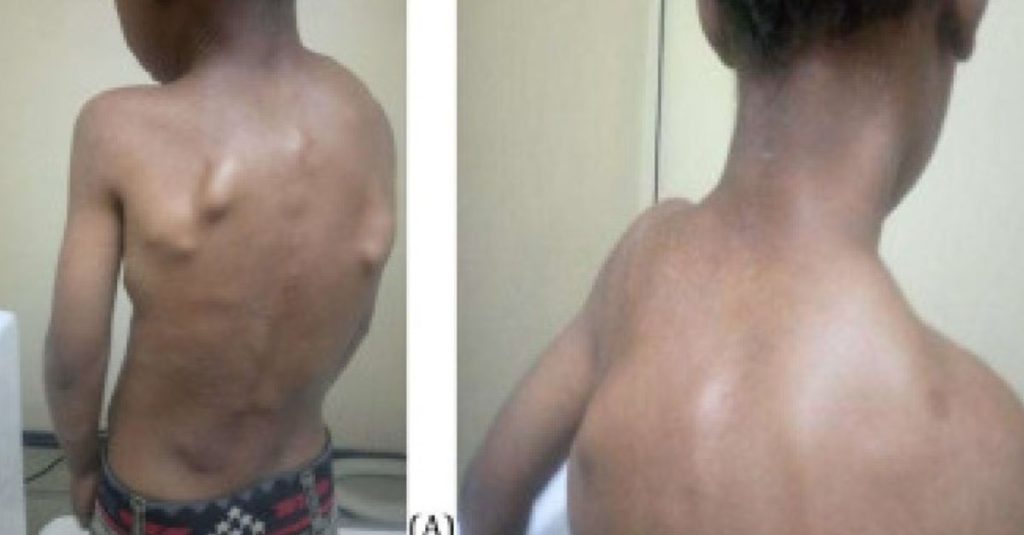
Our first patient is a 9-year-old boy who presented with multiple swellings across his neck and back. These swellings had persisted for 5 years before his arrival. He was born from non-consanguineous parents following an uneventful term pregnancy with no familial history of similar symptoms or conditions.
At the age of 4, he was initially misdiagnosed and managed as a case of multiple exostoses, a condition featuring bony projections capped by cartilage, commonly developing in the metaphysis of long bones. The patient received non-surgical treatment and was unfortunately lost to follow-up.
Five years later, the patient returned, reporting increased neck swelling, stiffness, and pain. Physical examination revealed several hard swellings at occipital area, the posterior, and lateral neck region, parascapular, and thoraco-lumbar paraspinal (Fig. 1).
Source: Science DirectRecent Posts
-
 A Week of Preventive Treatment and Emergency Flare-Up Management
A Week of Preventive Treatment and Emergency Flare-Up Management -
 Mayeshree’s Journey — Staying Ahead of Future Flare-Ups
Mayeshree’s Journey — Staying Ahead of Future Flare-Ups -
 Coordinating Hospitals and Doctors Across India for Seamless FOP Care
Coordinating Hospitals and Doctors Across India for Seamless FOP Care -
 FOP Awareness Expands — Training Program Held for Police Personnel
FOP Awareness Expands — Training Program Held for Police Personnel -
 Global Guidelines Continue to Shape Local Treatment Decisions
Global Guidelines Continue to Shape Local Treatment Decisions