Introduction
Heterotopic ossification (HO) is a disorder characterized by ectopic bone formation at extraskeletal sites, including skeletal muscle and connective tissues. Trauma-induced HO develops as a common post-operative complication after orthopedic surgeries (e.g., hip arthroplasty), blast injuries, deep burns and nervous system injuries (Hwang et al., 2022). In addition to trauma- induced HO, fibrodysplasia ossificans progressiva (FOP) is a rare congenital autosomal dominant disorder also involving HO (Bravenboer et al., 2015). Ectopic bones form progressively through endochondral ossification, mostly in episodic flare-ups associated with inflammation (Towler and Shore, 2022). These cumulative osteogenic events lead to reduced mobility resulting from ankylosing joints. Affected individuals have a shorter life-span, most commonly due to thoracic insufficiency syndrome (Kaplan et al., 2010; Pignolo et al., 2016).
HO reflects a shift from a normal tissue repair process to an aberrant reactivation of bone-forming developmental programs that require acute inflammation and the excessive expansion of local progenitors, followed by inappropriate differentiation of these progenitors into chondroblasts and osteoblasts which finally results in bone formation (Hwang et al., 2022). In both trauma- induced HO and FOP, pathology appears following the excessive activation of receptors sensitive to Transforming growth factor-β (TGFβ) superfamily family members (Lees-Shepard et al., 2018; Wang et al., 2018). FOP arises from gain-of-function mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1, with the most common mutation being c.617G>A, R206H (Shore et al., 2006). Higher SMAD 1/5 mediated signaling of mutated ACVR1/ALK2 has been partially attributed to a loss of auto-inhibition of the receptor and mild hypersensitivity to BMP ligands (Chaikuad et al., 2012; Groppe et al., 2011; Van Dinther et al., 2010). Signaling by mutated ACVR1 still relies on ligand-induced heterotetrameric clustering but, unlike wild type ACVR1, does not require ACVR2A/B kinase activity (Agnew et al., 2021; Ramachandran et al., 2021). More importantly, mutations alter their signaling specificity, abnormally transducing SMAD 1/5 and altering the non-canonical BMP signals (e.g., p38 or PI3K) in response to activin A (Hatsell et al., 2015; Hino et al., 2015; Valer et al., 2019a). Accordingly, evidence supports that activin A is necessary and sufficient for HO in FOP (Hatsell et al., 2015; Lees-Shepard et al., 2018; Upadhyay et al., 2017). In contrast, activin A does not drive post-traumatic HO (Hwang et al., 2020), and non-genetically-driven HO mostly arises from excessive BMP and TGFβ signaling, with functional redundancy between different type I receptors of the TGFβ-superfamily (Agarwal et al., 2017; Patel et al., 2022; Sorkin et al., 2020; Wang et al., 2018).
Evidence points to mesenchymal fibroadipogenic precursors (FAPs), which are widely distributed in muscle and other connective tissues, as the key cell-of-origin that aberrantly undergo chondrogenesis and further ectopic bone formation (Dey et al., 2016; Eisner et al., 2020; Lees- Shepard et al., 2018). However, either FOP or non-genetic HO also require local tissue destruction and inflammation, which indicate that FAPs should be invariably primed for ectopic bone formation by this inflammatory microenvironment (Barruet et al., 2018; Convente et al., 2018; Hwang et al., 2022; Matsuo et al., 2019; Pignolo et al., 2013; Sorkin et al., 2020).
Inflammation in HO is characterized by an initial acute response following injury with the activation of innate immunity and the influx of neutrophils and monocytes (Convente et al., 2018; Hwang et al., 2022). Additionally, B and T cells of the adaptive system are also recruited (Chakkalakal et al., 2012; Convente et al., 2018). However, mice lacking B or T lymphocytes exhibited no delay in the development of heterotopic ossification (HO) after injury, indicating that these cells may play a subsequent role in the dissemination of bone lesions (Kan et al., 2009). During intermediate and late inflammatory stages, the recruitment of monocytes, macrophages and mast cells occurs, which in turn exerts autocrine and paracrine effects on nearby FAPs (Chakkalakal et al., 2012; Convente et al., 2018; Sorkin et al., 2020; Tu et al., 2022). Highlighting its relevance in HO, the depletion of mast cells and macrophages profoundly impairs genetic and non-genetic HO (Convente et al., 2018; Torossian et al., 2017; Tu et al., 2022). BMP receptors are robustly expressed in monocytes and macrophages and the expression of ACVR1R206H has been shown to extend inflammatory responses in patient-derived macrophages (Barruet et al., 2018; Matsuo et al., 2021, 2019). From the plethora of cytokines secreted by monocytes, macrophages and mast cells, both activin A and TGF-β stand out as extremely relevant for HO (Alessi Wolken et al., 2017; Hatsell et al., 2015; Lees-Shepard et al., 2018; Patel et al., 2022; Sorkin et al., 2020; Upadhyay et al., 2017).
Genetic and pharmacological studies have indicated that osteochondroprogenitor specification and maturation depend on phosphatidylinositol 3-kinase-α (PI3Kα) (Ford-Hutchinson et al., 2007; Fujita et al., 2004; Gámez et al., 2016; Ikegami et al., 2011). We found that PI3K signaling was also linked to HO, since inhibitors of PI3Kα (BYL719/Alpelisib/Piqray) prevented HO in mouse models without major side-effects (Valer et al., 2019a). Mechanistically, PI3Kα inhibitors hamper canonical and non-canonical BMP signaling, decreasing total and phosphorylated SMAD1/5 levels and reducing transcriptional responsiveness to BMPs/Activin A in mesenchymal progenitors (Gámez et al., 2016; Valer et al., 2019b). In the current study, we demonstrate that the pharmacological and genetic inhibition of PI3Kα in HO progenitors at injury sites reduces HO in vivo. Moreover, envisioning a future translation into the clinic, we have optimized the administration of BYL719 and found that the delayed administration of BYL719, up to seven days after inflammatory injury, still prevents HO in mice. In addition, we found that BYL719 blocks osteochondroprogenitor specification and effectively reduces the essential inflammatory response. Altogether, the data presented here show the potent therapeutic effect of PI3Kα inhibition on HO in mouse models.
Results
Delayed initiation of treatment with PI3Kα inhibitor effectively prevents heterotopic ossification
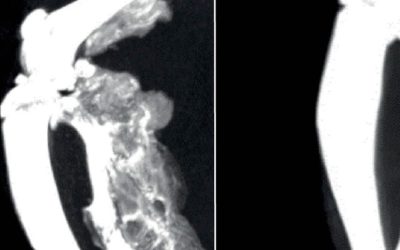
Heterotopic ossification takes place following a precise temporal pattern of progenitor activation and the recruitment of distinct cell types at the ossification centers which causes early divergence from normal skeletal muscle repair program. We previously found that the pharmacological administration of BYL719 prevents HO in a mouse model of HO (Valer et al., 2019a). Given that BYL719 is marketed for the treatment of cancer and overgrowth syndrome, it is a promising therapeutic molecule in pathological HO. To further determine a potential therapeutic window for BYL719 and better understand the cellular targets of BYL719, we evaluated the effect of BYL719 when administered intermittently and several days after HO induction. For this, we used a conditional mouse model (ACVR1Q207Dfl/fl) in which we combine the overexpression of Cre recombinase through adenoviral particles with the injection of cardiotoxin intramuscularly in the hindlimb (Fukuda et al., 2006). In this model, we administered BYL719 (i.p. 25 mg/kg) or vehicle control intermittently starting one, three or seven days after HO induction. We also implemented a different regimen involving the administration of BYL719 only for the initial three days following the injury (Figure 1A). None of the treatments led to significant changes in the weight of the mice (Figure 1-Figure Supplement 1). HO volume was analyzed by micro computed tomography (µCT) twenty-three days post-injury. The early administration of the treatment with BYL719 at day one or three after the onset of HO resulted in significantly lower HO; late administration seven days after the induction of HO was also partially effective (Figure 1B and C). Discontinuation of the treatment after three days did not prevent HO at day 23. These results indicate that the inhibition of PI3Kα by BYL719 after injury prevents HO and this protection is still effective if the treatment is started several days after injury, expanding its therapeutic window.
Deficiency of PI3Kα at injury sites is sufficient to partially prevent heterotopic ossification
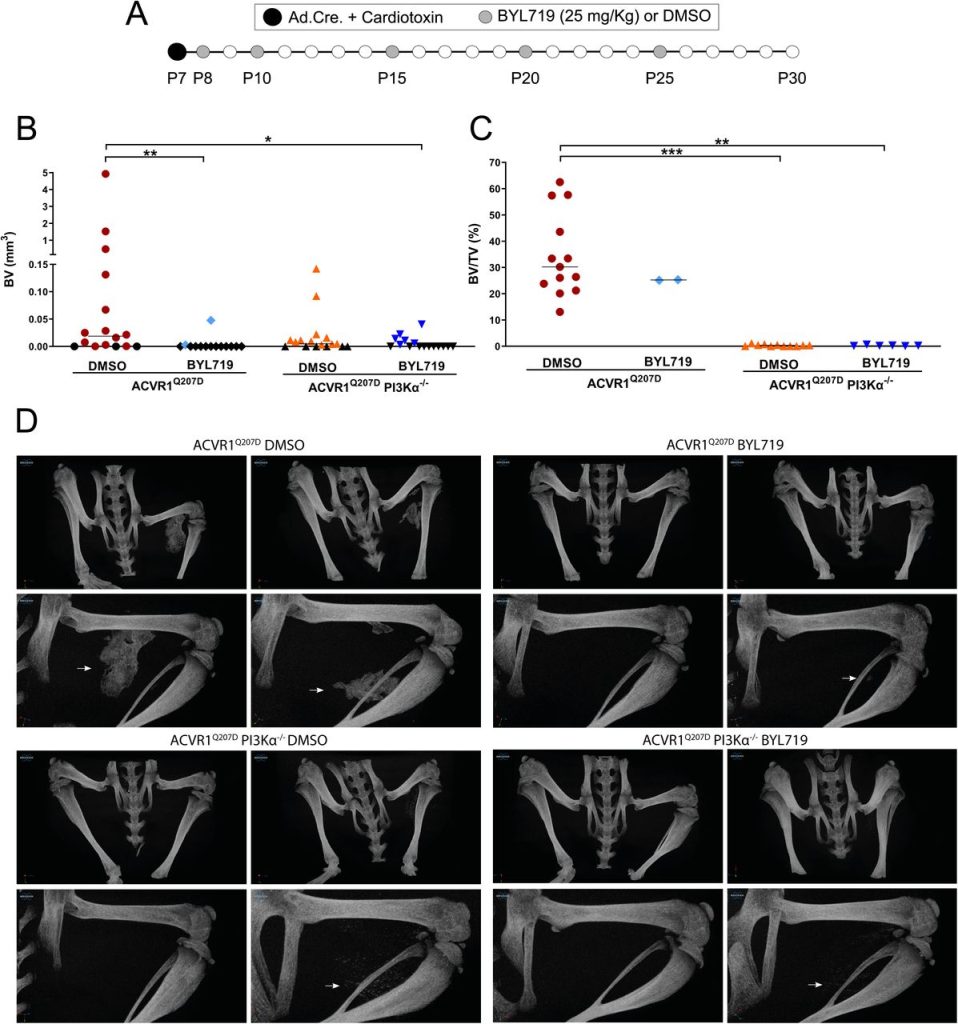
Since other small molecules designed to target PI3Kα have reported partial off-target effects on kinases other than PI3Kα (Furet et al., 2013; Jamieson et al., 2011), we aimed to confirm that the results obtained with BYL719 were due to the specific inhibition of PI3Kα. Therefore, we developed a conditional mouse model (ACVR1Q207Dfl/fl:p110αfl/fl) in which the Cre recombinase drives the expression of ACVR1Q207D and deletion of the catalytic subunit of PI3Kα (p110α) after intramuscular hindlimb combined injection of adenovirus-Cre and cardiotoxin to trigger HO. With this approach, Cre mediates the expression of ACVR1 Q207D and the deletion of PI3Kα in the same cells, whereas cells recruited afterwards would be wild type for both genes. As expected, µCT analysis performed 23 days after injury showed extensive HO in mice expressing ACVR1Q207D, which was prevented by intermittent treatment with BYL719 initiated one day after injury (Figure 2A, 2B and 2D). Genetic deletion of p110α in mice expressing ACVR1Q207D led to a reduction in HO, which was further enhanced by BYL719 (Figure 2B). Importantly, no significant changes in the weight of the mice were observed, confirming the absence of severe toxicity (Figure 2-Figure Supplement 1A). Histomorphometrically, ACVR1Q207D mice wild type for PI3Kα developed islands and/or bone spurs with a well-organized bone structure. However, in PI3Kα deficient mice ACVR1Q207D expression only led to minor ectopic calcifications that were already surrounded by fully regenerated muscle tissue on the 23rd day after injury (Figure 2D, Figure 2-Figure Supplement 1B). Accordingly, analysis of the ratio bone volume/tissue volume (BV/TV) and the correlation between bone volume and BV/TV of individual mice was clearly different between p110α genotypes, irrespective of BYL719 treatment (Figure 2C, Figure 2-Figure Supplement 1C). These results demonstrate that the abrogation of PI3Kα activity (either genetic of pharmacologically) in cells at injury sites is relevant for their inhibitory effects in HO. This might be attributed to the direct effects of BYL719 on progenitor cell expansion and chondroblast specification, as well as its possible ability to reduce the inflammatory response required for HO.
Recent Posts
-
 A Week of Preventive Treatment and Emergency Flare-Up Management
A Week of Preventive Treatment and Emergency Flare-Up Management -
 Mayeshree’s Journey — Staying Ahead of Future Flare-Ups
Mayeshree’s Journey — Staying Ahead of Future Flare-Ups -
 Coordinating Hospitals and Doctors Across India for Seamless FOP Care
Coordinating Hospitals and Doctors Across India for Seamless FOP Care -
 FOP Awareness Expands — Training Program Held for Police Personnel
FOP Awareness Expands — Training Program Held for Police Personnel -
 Global Guidelines Continue to Shape Local Treatment Decisions
Global Guidelines Continue to Shape Local Treatment Decisions